Ordered, immediately called back and the same day delivered the order.Very pleased with the work. Thank you for prompt and accurate work https://africarx.co.za/ great prices, delivered on the day of the order. Pleasant managers consult by phone.
Acdrupal.evergreen.edu
doi: 10.1146/annurev.med.54.101601.152421
Copyright c 2003 by Annual Reviews. All rights reserved
FUNCTIONAL GENOMICS OF THE PARAOXONASE (PON1) POLYMORPHISMS: Effects on Pesticide Sensitivity, Cardiovascular Disease, and Drug Metabolism
Lucio G. Costa,1 Toby B. Cole,1,2 Gail P. Jarvik,2 andClement E. Furlong2Departments of 1Environmental Health and 2Genome Sciences and Medicine, Divisionof Medical Genetics, University of Washington, Seattle, Washington 98195;e-mail: lgcosta@u.washington.edu; tobycole@u.washington.edu;pair@u.washington.edu; clem@u.washington.eduKey Words metabolism of oxidized lipids, organophosphate metabolism, linkage
disequilibrium, nerve agents, drug metabolism, statin metabolism
■ Abstract This review focuses on the functional genomics of the human paraox- onase (PON1) polymorphisms. Levels and genetic variability of the PON1 position 192 isoforms (Gln/Arg) influence sensitivity to specific insecticides or nerve agents and risk for cardiovascular disease. A more recent area of investigation, the role of PON1 in drug metabolism, is also discussed. We emphasize the importance of considering both PON1 isoforms and PON1 levels in disease/sensitivity association studies. INTRODUCTION
Paraoxonase-1 (PON1) is a high-density lipoprotein (HDL)–associated serum en-zyme whose primary physiological role is to protect low-density lipoproteins(LDL) from oxidative modifications (1). Early research focused on the obser-vation that PON1 could hydrolyze organophosphorus (OP) compounds, includingcommonly used insecticides (2). Indeed, the enzyme (EC 3.1.8.1) was initiallycharacterized as an organophosphate hydrolase, and its name derives from one ofits most commonly used in vitro substrates, paraoxon. More recently, in addition toits role in lipid metabolism and, hence, in cardiovascular disease and atherosclero-sis, PON1 has been shown to play a role in the metabolism of pharmaceutical drugs.
Early studies, which used the single substrate paraoxon to measure PON1
paraoxonase activity in serum from subjects of Caucasian origin, revealed a bi-modal or trimodal distribution (3–5). On the basis of enzymatic tests, humans couldbe divided into three serum PON1 phenotypes: those with low, intermediate, and
high PON1 activity, without a clear demarcation between intermediate and highmetabolizers. Studies in the past decade have elucidated the genetics of PON1and provided analyses that clearly differentiate among the PON1 phenotypes, asdescribed below. Two polymorphisms in the coding region (6), as well as five inthe promoter region, have been identified (7–11). The Q192R polymorphism de-termines catalytic efficiency of hydrolysis of some substrates (12–16), and someof the promoter polymorphisms, particularly C-108T, help regulate the level ofexpression of PON1 (7–11). This review focuses on the functional genomics ofthe PON1 polymorphisms with respect to their role in determining susceptibilitiesto the toxicity of certain pesticides and to cardiovascular disease, as well as theirrecently emerging role in drug metabolism. PON1 STRUCTURE AND POLYMORPHISMS
Studies in the early 1990s led to the purification of rabbit and human PON1s(6, 18) and subsequent cloning and sequencing of their respective cDNAs (6, 13). The PON1 cDNA encodes a protein of 355 amino acids from which only theamino-terminal methionine residue is removed during secretion and maturation(6). The retained leader sequence is required for the association of PON1 withHDL particles (19), and indeed PON1 is entirely associated with HDL in humanserum (20). PON1 protein is synthesized mostly in the liver, from which it isreleased by a docking process whereby HDL particles transiently associate withthe cell membrane and remove PON1 from the membrane (21). Physical mappingplaced the human PON1 gene on chromosome 7 q21–22 (14). Two polymorphismswere observed in the PON1 coding sequence: a Gln (Q)/Arg (R) substitution atposition 192 and a Leu (L)/Met (M) substitution at position 55 (6, 13, 14). Us-ing the PCR method described by Humbert et al. (14) or modifications thereof,many studies have established PON1 192 and 55 genotypes for individuals inlarge populations. The polymorphism at position 192 has been the most studied,with gene frequencies for PON1Q192 ranging from 0.75 for Caucasians of North-ern European origin to 0.3 for some Asian populations (11). On the other hand,the gene frequency for PON1L55 ranges from 0.57 in Caucasian populations to0.99 in an Oji-Cree population. Several studies have shown that the L55 and R192alleles are in strong disequilibrium, with ∼98% of the R192 alleles having L atposition 55 (11).
The coding region polymorphisms in the PON1 protein have been studied for
effects on the catalytic efficiencies of hydrolysis of specific substrates. The L/Mpolymorphism at position 55 has not been found to affect catalytic efficiency (13–16) but has been associated with the variability of plasma PON1 levels; PON1M55individuals have lower PON1 levels on average (10, 22, 23). Linkage disequilib-rium of the PON1M55 allele with promoter polymorphisms appears to explain mostof the differences observed (11), although PON1M55 has been reported to be lessstable than PON1L55 (24). On the other hand, the Q/R polymorphism of position
192 significantly affects the catalytic efficiency of PON1, in a substrate-dependentmanner (15). Initial studies indicated that the PON1R192 isozyme hydrolyzedparaoxon more readily than PON1Q192 (13, 14). Further studies suggested thatthe effects of this polymorphism may be substrate-dependent, as the PON1Q192isoform hydrolyzed diazoxon, sarin, and soman more rapidly than PON1R192 within vitro assays (15).
Additional polymorphisms have been found in the noncoding region of the
PON1 gene (7–11). These polymorphisms are at positions –108 (C/T), –126 (G/C),–162 (A/G), –832 (G/A), and –909 (C/G), although reports vary somewhat in as-signment of the base positions (10). The most significant of these promoter regionpolymorphisms is that at position –108, which contributes 22.4% of the variationin PON1 expression, with –108C providing higher levels of plasma PON1. Thepolymorphism at position –162 also contributes a small (2.4%) amount (10). Con-tribution of the 3 polymorphisms (10) to variability of PON1 expression has notyet been investigated.
The existence of PON1 coding-region polymorphisms, which affect catalytic
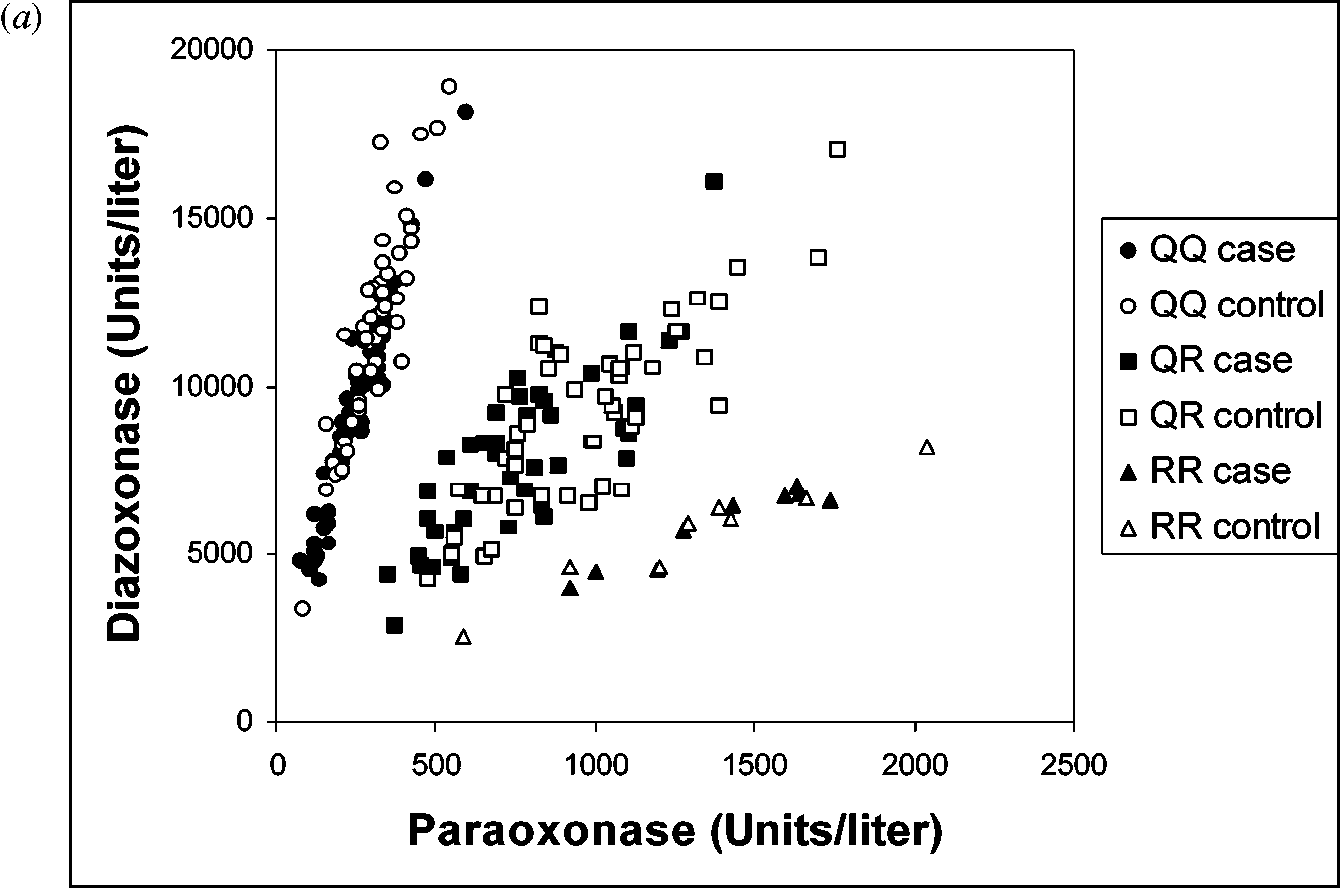
activity toward OPs, and of promoter-region polymorphisms, which affect thelevels of PON1 expression, led to the proposal of determining the “PON1 status”of an individual (25, 26). By plotting rates of diazoxon hydrolysis against paraoxonhydrolysis at high salt concentrations (2 M NaCl), an accurate inference of PON1192genotype, as well as PON1 activity levels for individuals, can indeed be made asshown in Figure 1 (15, 25–27). PON1 AND SENSITIVITY TO ORGANOPHOSPHATE TOXICITY
Many OP compounds are triesters of phosphoric acid. Their major source is frominsecticides, although some have found therapeutic applications. Acute exposureto OPs causes neurotoxicity, in the form of a cholinergic syndrome, i.e., an over-stimulation of muscarinic and nicotinic acetylcholine receptors in the central andperipheral nervous systems, due to accumulation of acetylcholine in the synapticcleft resulting from OPs’ inhibition of acetylcholinesterase (28).
Only OPs with a P=O moiety can interact with acetylcholinesterase. Commonly
used insecticides, such as diazinon and chlorpyrifos, that have a P=S bond, need tobe converted to their oxygen analogs. Bioactivation occurs by a process of oxidativedesulfuration catalyzed by cytochrome P450 enzymes. A-esterases, such as PON1,can hydrolyze, and hence detoxify, a number of insecticidal organophosphates,including some nerve agents (2, 15).
The existence of polymorphisms in PON1 that confer different hydrolyzing
ability toward OPs, as well as different levels of expression, has long inspiredthe hypothesis that certain individuals may be more sensitive to OP toxicity (29). This hypothesis has received confirmation only in the past decade, primarily fromanimal studies. Initial evidence for the role of PON1 in OP toxicity came from
cross-species comparisons, from animal experiments using purified PON1, andmore recently from studies with PON1 knockout mice. Earlier findings indicatedthat birds, which have no to very low plasma PON1 activity, were more sensitivethan rats to the toxicity of various OPs (30). In turn, rats were more sensitive to thetoxicity of OPs than rabbits, whose plasma PON1 activity is seven times higher(31). Although several other factors may contribute to the species differences inOP toxicity, these early findings suggested that low plasma PON1 activity wouldincrease sensitivity to the acute effects of OPs.
A more direct approach was provided by studies in which exogenous PON1
was injected into rats or mice. In a pioneering experiment, Main (29) intravenouslyinjected into rats a partially purified PON1 from rabbit serum and noted a decreasein the acute toxicity of paraoxon. More recent studies in rats and mice have con-firmed and expanded this early finding. Initially, PON1 purified to homogeneity
(a) Plot of diazoxonase vs. paraoxonase activities (26, 27) for carotid artery
disease (CAAD) cases and controls, coded for PON1192 genotypes (determined bypolymerase chain reaction). Note that the two-substrate assay provides an accurateinference of PON1192 genotype as well as the level of plasma PON1 activities (i.e.,PON1 status). Reproduced from Reference 27 with permission. (b) Diazoxonase levelsof CAAD patients and controls separated by position 192 genotypes as indicated. Notethe abundance of individuals with low diazoxonase among the PON1QQ cases andthe sparcity of cases with high diazoxonase activity. Diazoxonase activity is a relativemeasure of plasma PON1 protein. Means are indicated by horizontal bars. Reproducedfrom Reference 27 with permission.
from rabbit serum (31a) was given by intravenous injection to rats (32). Adminis-tration of the enzyme raised rat serum PON1 activity toward paraoxon and chlor-pyrifos oxon by 9- and 50-fold, respectively. When rats were challenged witheither of these OPs, a significant protection [assessed by measuring inhibition ofacetylcholinesterase (AchE) in different tissues] was observed, particularly againstchlorpyrifos oxon. The protection was more prominent in two target tissues (brainand diaphragm), and was also present when OP exposure occurred by the dermalroute, as is often the case for occupationally exposed workers (32).
Other experiments, which followed a similar protocol, extended these findings
to mice (25). Furthermore, it was found that serum PON1 levels could be increasedfor extended periods (t1/2 > 30 h) by administering purified PON1 intravenouslyand intramuscularly (25). Additional studies indicated that exogenous PON1 could
also afford protection against the toxicity of chlorpyrifos, the parent compoundused as an insecticide, when given before or even up to 3 h after OP exposure(33). Recently, recombinant human PON1 (either LR or LQ) expressed from anadenoviral vector was shown to increase serum paraoxonase activity by about60% and to protect mice against the toxicity of chlorpyrifos (34). Overall, thesestudies indicated that by artificially increasing serum levels of PON1 it is possibleto decrease the toxicity of certain OPs.
More recent experiments have investigated the toxicity of OPs in PON1 knock-
out (PON1−/−) mice, which were produced by targeted disruption of exon 1 ofthe PON1 gene (35). PON1−/− mice have no plasma or liver hydrolytic activitytoward paraoxon and diazoxon, and a very low level of activity toward chlorpyri-fos oxon (16). PON1 knockout mice have dramatically increased sensitivity tochlorpyrifos oxon and diazoxon and slightly increased sensitivity to the respectiveparent compounds chlorpyrifos and diazinon (16, 35). A surprising finding camefrom the experiments with paraoxon, the OP after which PON1 was named. PON1knockout mice did not show increased sensitivity to paraoxon, despite the totallack of paraoxonase activity in plasma and liver (16).
Further experiments investigated whether administration of purified PON1
would restore plasma (but not liver) PON1 levels, and thereby OP resistance, inPON1−/− mice. Either human PON1Q192 or PON1R192 was injected intravenouslyinto PON1−/− mice, and the effects of OPs on brain and diaphragm AChE weredetermined. In the case of chlorpyrifos oxon, both isoforms were protective, andPON1R192 offered about 50% better protection than PON1Q192 (16). Both PON1R192and PON1Q192 offered equal protection against diazoxon, and neither human PON1isoform protected against the toxicity of paraoxon, extending the surprising find-ings described above. The results from the kinetic analysis of substrate hydrolysisby purified human PON1192 isoforms provide an explanation for such findings. Although the PON1R192 isoform is eight times more efficient than the PON1Q192isoform in hydrolyzing paraoxon (Km/Vm = 6.27 versus Km/Vm = 0.71),neither isoform hydrolyzes paraoxon as efficiently as diazoxon or chlorpyrifosoxon (16). This confirms the hypothesis (36) that PON1 is not efficient at hy-drolyzing paraoxon at low concentrations, suggesting that PON1 may not degradeparaoxon efficiently in vivo and that other pathways (e.g., cytochromes P450, car-boxylesterases) are primarily responsible for detoxifying paraoxon in vivo (36, 37). The catalytic efficiency of PON1Q192 for diazoxon (Km/Vm = 75) was almostthe same as that of PON1R192 (Km/Vm = 77), and the catalytic efficiency ofPON1R192 for chlorpyrifos oxon was significantly higher than that of PON1Q192(Km/Vm = 256 versus Km/Vm = 150) (16).
In summary, this series of studies provided evidence that PON1, whose catalytic
activity and level of expression are determined by polymorphisms in the codingand 5 -regulatory regions, respectively, plays an important role in modulating thetoxicity of some, but not all, OPs, which are in vitro substrates for this enzyme. The catalytic efficiency of hydrolysis of oxons determines whether PON1 providesprotection against a specific OP exposure.
An additional issue related to the role of PON1 in determining sensitivity to
OP toxicity stems from the possibility that young children may be more sensitivethan adults to the toxic effects of certain pesticides (38). The acute toxicity of OPsappears to be influenced by age, with young animals being more sensitive (39, 40),although whether such enhanced sensitivity extends to situations of repeated sub-lethal exposures is still unclear (41). Furthermore, young animals have been foundespecially sensitive to the central neurotoxicity of certain OPs (42), whereas theyappear resistant to organophosphate-induced delayed polyneuropathy (43). Thereis an increasing consensus that lower metabolic abilities of young animals are amajor determinant of their increased sensitivity to OP toxicity, as suggested a fewdecades ago (39). In particular, studies with chlorpyrifos have concluded that alower hydrolytic detoxification by PON1, and perhaps carboxylesterase, accountsfor the differential age-related sensitivity in its acute toxicity (44, 45). Indeed, inboth rats and mice, liver and serum PON1 activity toward different substrates, aswell as liver PON1 mRNA, increases from birth to postnatal day 21 (46). Thereis also evidence of a low serum paraoxonase activity, measured with paraoxon (4)or phenylacetate (47, 48) as substrates, in human infants, which seems to reacha plateau (whose level is determined by PON1 regulatory-region polymorphismsand the genetic background of the individual) after one year of age (2, 49). PON1 AND CARDIOVASCULAR DISEASE
The influence of PON1 and its polymorphisms on cardiovascular disease appearsrelated to its ability to metabolize oxidized lipids and prevent their formation. Oxidized LDL is believed to play a central role in monocyte chemotaxis andmacrophage differentiation, which are early events in the progression of atheroscle-rosis, whereas HDL destroys these biologically active oxidized lipids (reviewedin Reference 50). PON1, the major enzyme responsible for this protective effect,is associated with the HDL particle (51). The HDL particle appears to acceptPON1 directly by high-affinity desorption as it is released from the liver, in a pro-cess that requires both apolipoprotein and phospholipids (21, 52). Mackness et al. (53) were the first to demonstrate that purified human PON1 could inhibit LDLoxidation in vitro. Other studies have confirmed and extended this finding, demon-strating that PON1 both prevents the formation of oxidized LDL and inactivatesLDL-derived oxidized phospholipids once they are formed (51, 54–59). PON1also protects phospholipids in HDL from oxidation (56). The lipid substrate speci-ficity of PON1 has not yet been fully defined. PON1 hydrolyzes phospholipid andcholesteryl ester hydroperoxides and hydroxides derived from arachidonic acid orlinoleic acid (54, 56–58). Purified PON1 can also reduce linoleic acid hydroperox-ides to linoleic acid hydroxides (58). Watson et al. (54) suggested that PON1 hy-drolysis of phospholipid hydroperoxides produces aldehyde or ketone derivativesthat then serve as substrates for platelet-activating factor acetylhydrolase (PAF-AH), generating lysophospholipids such as lysophosphatidyl choline. Ahmed et al.
(60) demonstrated that oxidized HDL lipids are converted by apolipoprotein A-1to phosphatidylcholine aldehydes that are subsequently hydrolyzed to lysophos-phatidylcholine by PON1, without detectable release of lineolate, arachidonate, ortheir hydroperoxy or hydroxy derivatives. Thus, PON1 appears to have esterase-like, peroxidase-like, and phospholipase-like activities that inhibit the formation ofproinflammatory oxidized phospholipids and degrade them once they are formed.
The strongest evidence for the connection between hydrolysis of oxidized lipids
by PON1 and increased susceptibility to atherosclerosis comes from a study ofPON1–/– mice. Mice without PON1 exhibited significantly larger aortic atheroscle-rotic lesions than wild-type mice when fed a high-fat diet, and HDL from thesemice failed to protect against LDL oxidation and monocyte chemotaxis in an invitro co-culture model of the artery wall (35). Thus, mice lacking PON1 were moresusceptible than wild-type mice to both lipoprotein oxidation and atherosclerosis.
Other studies in mice, humans, and other species demonstrated that low plasma
PON1 activity is associated with features of atherosclerosis. In apolipoprotein E(ApoE) knockout mice, LDL receptor knockout mice, and apoAII-overexpressingtransgenic mice, plasma PON1 levels are greatly reduced and the mice exhibit hy-percholesterolemia and atherosclerosis (61, 62). Furthermore, adding PON1 backto HDL from apoAII transgenic mice restored the protective ability of HDL againstlipid hydroperoxide formation (62). HDL isolated from normolipidemic patientswith low plasma PON1 activity was unable to protect against LDL lipid oxidation(61). Avian HDL is also deficient in PON1 and unable to prevent LDL oxidation(63). In mice lacking both PON1 and ApoE, very-low-density lipoprotein (VLDL),intermediate-density lipoprotein (IDL), and LDL contained more oxidized lipidthan did VLDL, IDL, or LDL isolated from mice lacking solely ApoE (64). How-ever, clearance of LDL was more rapid in PON1−/− mice. Shih et al. (65) attributedthis to increased LDL oxidation, leading to formation of LDL-immunoglobulincomplexes that can be removed by the liver. An atherogenic diet decreases plasmaPON1 activity (61, 66, 67), at least partly due to changes in liver PON1 transcription(66). Van Lenten et al. (68) showed that oxidized 1-palmitoyl-2-arachadonoyl-sn-glycero-3-phosphorylcholine (ox-PAPC) reduces hepatic PON1 expression via aninterleukin (IL)–6-mediated pathway.
Based on the studies described above on the role of PON1 in OP detoxification,
it is likely that variability among individuals in the PON1-catalyzed hydrolysisof oxidized lipids arises from two sources. The first source of variability is thepolymorphism at PON1Q192R, which results in different, substrate-specific catalyticefficiencies of hydrolysis for PON1Q192 and PON1R192. The frequent associationof PON1R192 with vascular disease predicts that this isoform would have a lowercatalytic efficiency of hydrolysis of oxidized lipids than PON1Q192. Indeed, this hasbeen shown to be the case experimentally (55, 57, 58, 69). Aviram et al. (58) foundthat both isoforms reduced total lipid-peroxide content of human atheroscleroticlesion homogenates, with PON1Q192 being twice as efficient as PON1R192. Thesecond source of variability in PON1 activity is PON1 abundance in the plasma,which has been largely ignored in studies examining the association of PON1
with atherosclerosis or other diseases. The magnitude of individual variability inplasma PON1 levels (up to 13-fold among individuals of the same PON1Q192Rgenotype) is probably why PON1Q192R genotype alone is not a sufficient predictorof disease (26). Because determining both PON1Q192R genotype and plasma PON1levels gives a more accurate measure of total PON1 activity, we and others haveemphasized strongly the importance of obtaining both measures, i.e., determiningan individual’s “PON status” (25–27, 70).
As described below, hydrolysis of the cholesterol-reducing drugs lovastatin,
mevastatin, and simvastatin by PON1, and altered paraoxonase activity associatedwith statin drug treatment, add additional layers of complexity to the involvement ofPON1 in the progression and treatment of atherosclerosis. Two other paraoxonasefamily members, PON2 and PON3, are capable of both preventing the formationof mildly oxidized LDL and inactivating mildly oxidized LDL once it is formed(71, 72). Whereas PON3 is produced in the liver and is associated with the HDLparticle, PON2 is ubiquitously expressed and does not appear to be part of HDL. It will be important to determine the relative contributions of each of these PONfamily members to atherosclerosis.
Studies of PON1 genotype as a predictor of human vascular disease have re-
ported mixed results, but overall they suggest that genotype is one of many pre-dictors of vascular disease. The PON1192R allele or PON1192RR genotype havebeen associated with cardiovascular disease in many (73–80), but not all (81–90),studies. Table 1 summarizes the simple odds ratios derived from these associationstudies.
Although the results of PON1192 genotype-disease association studies are mixed,
when a PON1192 genotype association with vascular disease was significant, it wasconsistently the PON1192R allele that was associated with disease. This finding in-dicates that the association is a true, but weak or variable, positive. It is likely thatthe studies’ results are mixed because of the heterogeneity among the populationsstudied. There may be more power to detect PON1192 genotype effects in stud-ies with Japanese cohorts owing to their higher PON1192R allele frequency (91),despite their lower incidence of vascular disease.
The PON1192R and PON155L alleles are in strong linkage disequilibrium in
multiple ethnic groups (22, 92–93). The PON155LL genotype predicted CHD inseveral studies, including a Caucasian sample in which PON1192 genotype did notpredict disease (94). PON155 genotype did not predict cardiovascular disease inother studied cohorts (70, 77, 88, 95), including an Asian Indian sample in whichPON1192 genotype did predict cardiovascular disease (92). Newer data suggestthat the PON1L55M polymorphism is correlated with the plasma PON1 concentra-tion both through linkage disequilibrium with promoter polymorphisms (10) andpossibly through increased protein turnover of the PON1M55 isoform (24).
PON1 activity level appears to be a better predictor of vascular disease than
genotype. In a sample of 212 predominantly Caucasian men, we found low-ered PON1 activity in subjects with severe carotid artery disease versus age-and race-matched controls (27). PON1 activity predicted carotid artery disease
Odds ratiosa from previous studies of the PON1192 polymorphism and
controls Outcomec Studies reporting a positive association Studies reporting no association
aOdds ratios for RR or QR genotype subjects compared with QQ genotype subjects where not reported werecalculated from the available data.
bSome rounding errors were necessary when the exact numbers of individuals with each genotype were not given,because they had to be reconstructed from genotype proportions.
cAbbreviations: CHD, coronary heart disease; CAD, coronary artery disease; MI, myocardial infarction. dEntire sample has type 2 diabetes. eOR for RR + QR vs. QQ.
independently of traditional risk factors. PON1192 ( p = 0.75) and PON155 ( p =0.83) genotypes or haplotype ( p = 0.70) did not predict case-control status unlessthe activity phenotype was also included as a predictor using logistic regression. This result suggested that the lowered PON1 activity earlier reported in 50 my-ocardial infarction survivors (95) was a risk factor, rather than a result, of theinfarction. The earlier study also failed to detect a significant marginal genotypeeffect, although the sample was small and, indeed, the odds ratio of the PON1192QRor PON1192RR genotype versus the PON1192QQ genotype was 2.42 (Table 1). Twogroups have confirmed an association between lowered PON1 activity and coro-nary heart disease (CHD) in addition to carotid artery disease (70, 96). Jameset al. (96) found that PON1 activity in type 2 diabetic patients was lower in those
with CHD than in those without CHD. In a study of 417 cases and 282 controls,Mackness et al. (70) detected significantly lowered PON1 activity in CHD cases.
Like Jarvik et al. (27), Mackness et al. (70) found lowered activity in vascular
disease with a marginal association of the PON1192 and PON155 coding-regionpolymorphisms (Table 1). Jarvik et al. (97) subsequently found that PON1–108 andPON1–162 5 promoter-region polymorphisms, like the coding-region polymor-phisms, did not predict case-control status. James et al. (96) reported an associa-tion of CHD with the lowered-expression, PON1–108TT genotype as well as with thePON155 polymorphism, which was in linkage disequilibrium with the PON1–108site. They did not detect a significant marginal effect of PON1192 on CHD in thetype 2 diabetics, although the odds ratios for the R-containing genotypes wereabove 1 (Table 1).
Mackness et al. (70) found lowered plasma PON1 levels associated with low-
ered PON1 activity in the cases. This suggests that it is the quantity of the enzyme,not its function, that is reduced. Surprisingly, James et al. (96) did not detect a sig-nificant decrease in PON1 concentration in cases, despite the significant PON1–107promoter-region polymorphism effect. However, the trend was toward lower con-centration in the cases.
Although PON1 activity is a better predictor of disease than genotype, PON1
genotypes remain important in the study of PON1’s role in vascular disease. Carefulstudies are needed to delineate the roles of both genetic and environmental influ-ences on PON1 activity. Gene-by-environment and gene-by-gene interactions canbe expected and will require large sample sizes to detect. One useful method ofexamining gene-by-gene interactions uses an elegant mouse genetic/proteomic ap-proach (98). Despite the significant linkage disequilibrium across the PON1 gene(10), few recent association studies have considered haplotype as a predictor ofdisease. Because of the common variants and the strong linkage disequilibrium,PON1 is amenable to newer methods to assign haplotypes in unrelated individu-als (99–101). Haplotype analyses will allow future analyses to consider the jointeffects of typed and untyped polymorphic sites. However, PON1 does present thechallenge of multiple functional sites leading to a large number of haplotypes. Appropriate haplotype groupings will be essential to improving power to detecthaplotype by disease association. Simultaneous consideration of PON1 activity,haplotypes, and environmental covariates will continue to elucidate the role ofPON1 in vascular disease. However, at this point in time, the two-substrate as-say (diazoxon and paraoxon) for determining PON1 status is the most informativeapproach for examining the relationship between PON1 status and disease (26, 27). PON1 AND DRUG METABOLISM
In addition to the oxygen analogs of some OP insecticides, the most commonly usedsubstrate for PON1 has been phenylacetate, which, however, is not a “polymorphicsubstrate” for the Q192R polymorphism. Recently, the ability of paraoxonase to
hydrolyze some novel substrates, most notably lactones and carbonate esters, hasbeen reported (102, 103). This finding may have several important consequences.
First, PON1 may be involved in the metabolism of pharmaceutical drugs, in
which case the PON1 status of an individual would be relevant to a drug’s effec-tiveness and/or side effects. Billecke et al. (103) found that the diuretic spirono-lactone and the hypocholesterolemic drugs mevastatin, lovastatin, and simvastatinare hydrolyzed by PON1, and that the Q and R isoforms hydrolyze these drugswith approximately the same efficiency. Pravastatin was found to increase serumapolipoprotein A1 and HDL cholesterol to a greater extent in men with the RRgenotype of PON1 than in those with the QQ genotype (104). Simvastatin therapyalso increases plasma paraoxonase activity (105), possibly by increasing the tran-scriptional activity of the PON1 gene (106). The therapeutic response of PON1activity is independent of PON1192 and PON155 polymorphisms (105).
Second, novel classes of so-called pro-drugs may be developed by incorporating
a lactone or cyclic carbonate moiety into other molecules, which can thus beinactivated or bioactivated in vivo by PON1. Inactivation has been shown to occurwith glucocorticoid γ -lactones (102) and activation with the antibacterial pro-drug prulifloxacin (107). The latter is hydrolyzed at a higher rate by type R thanby type Q serum. Interestingly, the free cysteine in position 284 of PON1, whichis not required for the hydrolysis of paraoxon or phenylacetate, is essential for thelactonase activity of the enzyme (103), as it is essential to protect LDL againstoxidation (57).
Third, PON1 may exert other effects on endogenous substrates, in addition to
protecting LDL from oxidation (57). It has been proposed that PON1 protectsagainst homocysteinylation by hydrolyzing homocysteine thiolactone, but the cat-alytic efficiency for this reaction is low (108). Although the Q isozyme is moreefficient in protecting against LDL oxidation, the R type hydrolyzes thiolactonesmore readily (103).
Finally, as noted above, two very toxic organophosphates, paraoxon and di-
azoxon, are currently used to determine PON1 status (26). The identification ofnovel PON1 substrates of low or no toxicity, which, unlike phenylacetate, are dif-ferentially hydrolyzed by the 192 R and Q alleles, will simplify the application ofthe two-dimensional assay to the study of large populations. One such compoundmay be the carbonate ester KB-R4899, which, like paraoxon, is hydrolyzed muchmore rapidly by the R than the Q isozyme of PON1 (103). MODULATION OF PON1 BY DIETARY, LIFESTYLE, AND ENVIRONMENTAL FACTORS
As discussed above, polymorphisms in the 5 regulatory region and coding regionsof PON1 determine its level of expression and enzymatic activity toward differentsubstrates. In humans, PON1 serum arylesterase activity increases from birth to 15–25 months of age, when it seems to reach a plateau whose level is determined by the5 regulatory-region polymorphisms and the genetic background of the individual
(2). In the adult, PON1 levels are stable and no significant changes have beenobserved with age (109, 110). However, efficient PON1 regulatory regions do notalone guarantee a high PON1 activity level (10). Enzyme inducers, environmentalchemicals, physiological and pathological states, and dietary and lifestyle factorshave demonstrated effects on PON1 activity.
Phenobarbital, a classical enzyme inducer, increases paraoxonase activity and
mRNA levels in rodent liver but not in serum (2, 111, 112). 3-methylcholanthrenewas found to increase both serum and liver PON1 activity in rats (113) but notin mice (2). Administration of lipopolysaccharide, which mimics gram-negativeinfections, causes a transient decrease in serum and liver PON1 activity and inhepatic mRNA levels (2, 114).
PON1 activity depends on calcium, and the calcium chelator EDTA abolishes
its activity. Other cations, however, have shown an inhibitory effect on PON1activity. Barium, lanthanum, copper, zinc, and mercurials were found to inhibitPON1 activity from rat or human liver in vitro (115). In vitro experiments withpurified human enzymes showed that PON1R192 was more sensitive than PON1Q192to inhibition by cadmium, zinc, mercury chloride, and iron, whereas PON1Q192displayed a higher sensitivity to inhibition by lead (116). In vivo, however, whenmice were treated with cadmium or methylmercury to achieve environmentallyrelevant blood concentrations, no inhibition of PON1 activity was found (116).
PON1 activity can vary depending on physiological conditions or pathological
states. For example, serum PON1 activity is significantly decreased during preg-nancy (5, 117). Low PON1 activity has been found in renal disease (118), diabetesmellitus (23, 119), various HDL deficiencies (120), and liver cirrhosis (121).
Dietary and lifestyle factors can also affect PON1 activity. Smoking has been
shown to decrease serum PON1 levels and activity (27, 122–124), both of whichappear to normalize relatively soon after cessation. In vitro experiments foundthat inhibition of PON1 activity by a cigarette-smoke extract was antagonized byreduced glutathione (GSH), N-acetylcysteine, and 2-mercaptoethanol, suggestingthat free thiols are central to the inhibitory effects (122). Ethanol and other aliphaticalcohols have been shown to inhibit serum PON1 activity (125); however, a studyin middle-aged men indicated that daily moderate alcohol consumption increasedserum PON1 activity, with no differences between wine, beer, and spirits (126). This increase may be due to the consumption of alcohol itself or to that of an-tioxidants, as similar results were obtained after consumption of red wine (127) orpomegranate juice (128, 129). A high-fat diet was shown to reduce serum PON1levels in mice (66, 67); in rats, dietary supplementation with fish oil also decreasedserum PON1, whereas supplementation with the triglyceride triolein increasedPON1 activity (130). Meals rich in thermally stressed olive oil, but not saffloweroil, were found to increase postprandial serum PON1 activity in middle-agedwomen but not in men (131).
The hypocholesterolemic drug simvastatin was found to increase serum PON1
activity (105, 106). As discussed above, statins and other lactones are hydrolyzedby PON1. On the other hand, the lactams, isosteric forms of lactones in which thering oxygen is replaced by nitrogen, inhibit PON1. Compounds such as
δ-valerolactam or ε-caprolactam inhibit PON1 activity, with IC50s in the mi-cromolar range (103).
These limited studies suggest that PON1 levels and activity can be modulated
by dietary, lifestyle, and possibly environmental factors. Although individuals withinefficient regulatory regions appear unable to express very high levels of PON1(10), the contribution of exogenous factors to an individual’s PON1 status shouldnot be discounted. CONCLUSIONS AND FUTURE PROSPECTS
Polymorphisms in the PON1 gene influence both the quantity and quality of PON1(i.e., PON1 status) (16, 26, 27). The quality of PON1 is governed by the Q192Rpolymorphism, which determines substrate-dependent differences in the catalyticefficiency of hydrolysis of specific OP substrates, including insecticide metabolitesand several nerve agents. Recent studies indicate that the Q192R polymorphismalso governs the efficiency of detoxification of biologically active oxidized lipids(54), and hence, the risk for vascular disease (27, 70, 96). Recent data on the roleof PON1 in the metabolism of specific drugs also indicate that PON1 status isimportant in determining the pharmacokinetics of metabolism (102, 103, 107).
The solid conclusion from all of these studies is that PON1 levels in all cases
and the Q192R polymorphism in some cases determine the rates at which a givenindividual will detoxify a specific insecticide, metabolize harmful oxidized lipids,and activate or inactivate specific drugs.
Interesting questions remain. It is clear that the 5 regulatory-region polymor-
phisms influence but do not fully determine PON1 levels. Other as-yet-unidentifiedgenes must also contribute to the large variability of plasma PON1 levels ob-served among individuals (15, 26, 27). These influences are often referred to asgenetic background contributions. The recent mouse proteomic research describedby Klose et al. (98) points to a direction for elucidating these influences. Kloseet al. were able to map mouse genes that influenced the levels of specific proteins.
The alignment of the three PON1 genes, PON1, PON3, and PON2, on chro-
mosome 7 provides an excellent system for examining questions of linkage dis-equilibrium in the human genome. Considerable linkage disequilibrium with thePON1 gene has already been described (10). It will be interesting to determinehow far outside the PON1 gene the blocks of linkage disequilibrium extend.
Last, but not least, the therapeutic possibilities of PON1, e.g., in treating in-
dividuals for exposure to nerve agents or insecticides or for reducing the riskof vascular disease, need to be examined. The animal studies described above(2, 16, 25, 33, 35) indicate that the injection of recombinant PON1 will providean efficient catalytic agent for detoxifying specific compounds in humans. Thestudies on the importance of catalytic efficiency (16, 36, 37) provide guidelines forengineering recombinant PON1 variants with catalytic efficiencies that would beefficacious for treating exposure to specific OP compounds.
Following the 1995 sarin releases in Japan, we determined a population dis-
tribution of hydrolysis rates for sarin and soman (15). The data showed that, likethe rates of hydrolysis of some OP insecticide metabolites, the rates of agenthydrolysis were significantly affected by the PON1R192Q polymorphism. Plasmafrom PON1R192 homozygotes demonstrated very low rates of sarin hydrolysis,whereas plasma from PON1Q192 homozygotes exhibited much better rates of sarinhydrolysis, depending on the level of PON1 present in the plasma. Heterozygotesexhibited intermediate rates of sarin hydrolysis. The rates of soman hydrolysiswere also somewhat higher for the PON1Q192 homozygotes than for the PON1R192homozygotes.
These observations led to the assumption that individuals with higher rates of
agent hydrolysis might be more resistant to exposure than individuals with lowrates of hydrolysis, i.e., PON1Q192 homozygotes might be more resistant to sarinexposure than PON1R192 homozygotes. Following our studies on the importanceof catalytic efficiency in providing protection against dermal exposures (16), weconcluded that the catalytic efficiency of agent hydrolysis by PON1 is probablynot adequate for significant protection against nerve agent exposure, in contrastto protection against diazoxon or chlorpyrifos oxon exposures. This conclusion isconsistent with the studies by Yamada et al. (132) on the PON1192 genotypes ofthe individuals exposed in the Tokyo subway sarin attack. Based on the currentstate of knowledge about PON1 status and possible resistance to nerve agentexposure, it would not make sense to screen plasma from individuals for high orlow rates of agent hydrolysis. For PON1 to be effective in treating agent exposure,recombinant PON1 with higher catalytic efficiencies of agent hydrolysis will needto be engineered. ACKNOWLEDGMENTS
Research by the authors was supported by grants from the National Institutesof Health and the Environmental Protection Agency (ES-04696, ES-09883, ES-11387, ES-07033, ES-09601/EPA-R826886, U19 ES11387). The Annual Review of Medicine is online at http://med.annualreviews.org LITERATURE CITED sic and Clinical Aspects. Norwell, MA:
rosis. Arterioscler. Thromb. Vasc. Biol.
2. Costa LG, Li WF, Richter RJ, et al. 2002.
arylesterase polymorphism. Am. J. Hum.
2a. Costa LG, Furlong CE. 2002. Paraox-onase (PON1) in Health and Disease: Ba-
family, biochemical and linkage studies.
15. Davies HG, Richter RJ, Keifer M, et al.
with diazoxon, soman and sarin. Nat.
onase polymorphism and specificity. Tox-
16. Li WF, Costa LG, Richter RJ, et al.
6. Hassett C, Richter RJ, Humbert R, et al.
2000. Catalytic efficiency determines the
in vivo efficacy of PON1 for detoxify-
ing organophosphates. Pharmacogenetics
onase: the mature protein retains its signal
sequence. Biochemistry 30:10141–49
7. Leviev I, James RW. 2000. Promoter poly-
and concentrations. Arterioscler. Thromb.
one esterase catalyzing both activities.
8. Suehiro T, Nakamura T, Inoue M, et al.
its association with PON1 expression.
apolipoprotein A-1 stabilizes activity. Arterioscler. Thromb. Vasc. Biol. 19:
20. Blatter MC, James RW, Messmer S, et al.
JB, et al. 2001. Effects of 5 regulatory-
fined by a lipoprotein-associated protein,
gene (PON1) expression. Am. J. Hum.
K-45. Identity of K-45 with paraoxonase.
11. Brophy VH, Jarvik GP, Furlong CE. 2002.
21. Deakin S, Leviev I, Gomaraschi M, et al.
PON1 polymorphisms. See Ref. 2a, pp.
12. Smolen A, Eckerson HW, Gan K, et al.
affinity, saturable, desorption mechanism.
serum paraoxonase/arylesterase. Drug.
22. Blatter Garin MC, James RW, Dussoix P,
13. Adkins S, Gan KN, Mody M, et al. 1993.
arylesterase: glutamine or arginine at po-
gene and increased risk of cardiovascular
sition 191, for the respective A or B al-
disease in diabetes. J. Clin. Invest. 99:62–
lozymes. Am. J. Hum. Genet. 53:598–608
14. Humbert R, Adler DA, Disteche CM, et al.
23. Mackness B, Mackness MI, Arrol S, et al.
dependent diabetes mellitus. Atheroscle-
24. Leviev I, Deakin S, James RW. 2001. De-
oxon toxicity in rats. Toxicol. Appl. Phar-
33. Li WF, Furlong CE, Costa LG. 1995.
concentrations. J. Lipid Res. 42(4):528–
fos toxicity in mice. Toxicol. Lett. 76:219–
25. Li WF, Costa LG, Furlong CE. 1993.
34. Cowan J, Sinton CM, Varley AW, et al.
phates. J. Toxicol. Environ. Health 40:
phosphate intoxication. Toxicol. Appl.
26. Richter RJ, Furlong CE. 1999. Determi-
35. Shih DM, Gu L, Xia YR, et al. 1998.
quires more than genotyping. Pharmaco-
atherosclerosis. Nature 394:284–98
27. Jarvik GP, Rozek LS, Brophy VH, et al.
36. Pond AL, Chambers HW, Chambers JE.
a better predictor of vascular disease than
is PON1192 or PON155 genotype. Arte-
esterases and aliesterase activities. Toxi-rioscler. Thromb. Vasc. Biol. 20:2441–47
37. Chambers JE, Ma T, Boone JS, et al. 1994.
pounds. In Experimental and ClinicalNeurotoxicology, ed. PS Spencer, H
Schaumburg, AC Ludolph, pp. 898–925.
secticides in the rat. Life Sci. 54:1357–
29. Main AR. 1956. The role of A-esterase
38. Eskenazi B, Bradnam A, Castorina R.
and parathion. Can. J. Biochem. Physiol.
adverse health effects. Environ. Health.
30. Brealey CB, Walker CH, Baldwin BC.
1980. A-Esterase activities in relation to
39. Benke GM, Murphy SD. 1975. The influ-
ence of age in the toxicity and metabolism
methyl to birds and mammals. Pestic. Sci.
and female rats. Toxicol. Appl. Pharma-
et al. 1987. Species differences in serum
40. Pope CN, Liu J. 1997. Age-related differ-
paraoxonase correlate with sensitivity to
paraoxon toxicity. In Toxicology of Pes-
pesticides. Environ. Toxicol. Pharmacol.ticides: Experimental, Clinical and Reg-ulatory Perspectives, ed. LG Costa, CL
41. Sheets LP. 2000. A consideration of age-
dependent differences in susceptibility to
42. Dam K, Seidler FJ, Slotkin TA. 2000.
human serum paraoxonase. Biochemistry
tion skills and locomotor activity. Dev.
54. Watson AD, Berliner JA, Hama SY, et al.
lipoprotein associated paraoxonase. Inhi-
bition of the biological activity of mini-
studies in developing chicks. Biochem.
mally oxidized low density lipoprotein. J.
55. Mackness B, Mackness MI, Arrol S, et al.
tribute to age-related sensitivity to chlor-
on the protection by high density lipopro-
pyrifos. J. Biochem. Toxicol. 11:279–87
tein against low density lipoprotein oxida-
45. Padilla S, Buzzard J, Moser VC. 2000.
tive modification. FEBS Lett. 423:57–60
the differential age-related sensitivity to
chlorpyrifos and metamidophos. Neuro-
serves its functions. A possible peroxida-
46. Li WF, Mathews C, Disteche CM, et al.
tive role for paraoxonase. J. Clin. Invest.
57. Aviram M, Billecke S, Sorenson R, et al.
and developmental expression. Pharma-
variation in plasma arylesterase activity
in children. Clin. Chim. Acta 8:568–73
arylesterase/paraoxonase activities: selec-
48. Augustinsson KB, Brody S. 1962. Plasma
lozymes Q and R. Arterioscler. Thromb.
born infants. Clin. Chim. Acta 7:560–65
49. Ecobichon DJ, Stephens DS. 1972. Peri-
58. Aviram M, Hardak E, Vaya J, et al. 2000.
terases. Clin. Pharmacol. Ther. 14:41–
50. Lusis AJ. 2000. Atherosclerosis. Nature
and peroxidase-like activities. Circulation
lipoprotein against oxidative modification
lipoprotein inhibits three steps in the for-
paraoxonase. Atherosclerosis 104:129–35
52. Noto H, Hashimoto Y, Satoh H, et al.
lipoprotein: steps 2 and 3. J. Lipid Res.
ciency. Biochem. Biophys. Res. Commun.
53. Mackness MI, Arrol S, Durrington PN.
lipoprotein oxidation with a peroxynitrite
donor. J. Biol. Chem. 276:24473–81
heart disease: Are activity and concentra-
tion more important than genotype? Arte-rioscler. Thromb. Vasc. Biol. 21:1451–57
71. Reddy ST, Wadleigh DJ, Grijalva V, et al.
ase ratio. J. Clin. Invest. 99:2005–19
et al. 1997. Overexpression of apolipopro-
tein AII in transgenic mice converts high
is not regulated by oxidized lipids. Arte-rioscler. Thromb. Vasc. Biol. 21:542–47
particles. J. Clin. Invest. 100:464–74
tously expressed protein with antioxidant
HDL. Biochem. Biophys. Res. Commun.
low density lipoprotein. J. Biol. Chem.
64. Shih DM, Xia YR, Wang XP, et al. 2000.
73. Ruiz J, Blanche H, James RW, et al. 1995.
hibit increased lipoprotein oxidation and
atherosclerosis. J. Biol. Chem. 275:
65. Shih DM, Reddy S, Lusis AJ. 2002. CHO
ical studies and transgenic mouse models.
coronary artery disease. J. Clin. Invest.
66. Shih DM, Gu L, Hama S, et al. 1996.
75. Pfohl M, Koch M, Enderle MD, et al.
atherogenesis in a mouse model. J. Clin.
67. Hedrick CC, Hassan K, Hough GP, et al.
76. Zama T, Murata M, Matsubara Y, et al.
by an immune mechanism. Arterioscler.
creased risk for coronary artery disease in
the Japanese. Arterioscler. Thromb. Vasc.
77. Sanghera D, Saha N, Aston CE, et al.
onase and the risk of coronary heart dis-
protein-1 via interleukin-6. J. Biol. Chem.
ease. Arterioscler. Thromb. Vasc. Biol.
78. Sanghera D, Aston CE, Saha N, et al.
associated with the risk of coronary heart
70. Mackness B, Davies GK, Turkie W, et al.
disease. Am. J. Hum. Genet. 62:36–44
79. Pati N, Pati U. 1998. Paraoxonase gene
ease in Indian subjects. Int. J. Cardiol.
artery disease. Pharmacogenetics 9:755–
80. Imai Y, Morita H, Kurihara H, et al.
phisms are not associated with cardiovas-
atherosclerotic diseases. Atherosclerosis
cular risk in renal transplant recipients.
90. Aubo C, Senti M, Marrugat J, et al.
cardiovascular events in elderly subjects.
82. Antikainen M, Murtomaki S, Syvanne M,
91. Odawara M, Tachi Y, Yamashita K. 1997.
risk of coronary artery disease in Finns. J.
dependent diabetes mellitus. Clin. En-
83. Rice G, Ossei-Gerning N, Stickland MH,
92. Sanghera D, Saha N, Kamboh MI. 1998.
heart disease. Coron. Artery Dis. 8:677–
with the risk of coronary heart disease in
84. Herrmann S, Blanc H, Poirier O, et al.
Asian Indians and Chinese. Atherosclero-
93. Boright A, Connelly PW, Brunt JH, et al.
Study. Atherosclerosis 126:299–303
and paraoxonase-2 is associated with vari-
heart disease. Int. J. Cardiol. 57:69–73
86. Ombres D, Pannitteri G, Montali A, et al.
carotid atherosclerosis: results of the Aus-
ated with coronary artery disease in Italian
trian Stroke Prevention Study. Stroke
patients. Arterioscler. Thromb. Vasc. Biol.
95. Ayub A, Mackness MI, Arrol S, et al.
87. Ko YK, Wang SM, Hsu LA, et al. 1998.
ocardial infarction. Arterioscler. Thromb.
96. James RW, Leviev I, Ruiz J, et al. 2000.
among Chinese in Taiwan. Atherosclero-
88. Cascorbi I, Laule M, Mrozikiewicz PM,
for coronary heart disease in type 2 dia-
betic patients. Diabetes 49:1390–93
97. Jarvik GP, Furlong CE, Hatsukami TS.
creased paraoxonase activity. J. Am. Coll.
(NM441), a prodrug of a new antibacterial
98. Klose J, Nock C, Hermann M, et al. 2002.
agent. Drug Metab. Dispos. 26:355–59
108. Jakubowski H. 2000. Calcium-dependent
et al. 1998. Haplotype structure and popu-
against protein N-homocysteinylation. J.
lation genetic inferences from nucleotide-
109. Playfer JR, Powell C, Evans DA. 1977.
lipase. Am. J. Hum. Genet. 63:595–612
Plasma paraoxonase activity in old age.
100. Fullerton SM, Clark AG, Weiss KM, et al.
2000. Apolipoprotein E variation at the se-
110. Zech R, Zurcher K. 1974. Organophos-
the origin and maintenance of a major hu-
ent mammals. Comp. Biochem. Physiol.
man polymorphism. Am. J. Hum. Genet.
111. Hernandez AF, Gonzalvo MC, Gil F, et al.
101. Stephens M, Smith NJ, Donnelly P. 2001.
1997. Divergent effects of classical induc-
A new statistical method for haplotype re-
construction from population data. Am. J.
tion paraoxonase and arylesterase. Envi-ron. Toxicol. Pharmacol. 3:83–86
112. Kaliste-Korhonen E, Tuovinen K, Han-
et al. 2000. Selective plasma hydrolysis of
glucocorticoid γ -lactones and cyclic car-
and γ -naphtoflavone on activities of dif-
ferent rat esterases after paraoxon expo-
ideal plasma inactivation mechanism. J.
103. Billecke S, Dragonov D, Counsell R, et al.
induction of paraoxonase in rat liver, kid-
cyclic carbonate esters. Drug Metab. Dis-
ney, lung and brain tissue. Implications for
its physiological role. Chem. Biol. Inter-
104. Malin R, Laaksonen R, Knuuti J, et al.
the effect of pravastatin on high-density
lipoprotein cholesterol. Pharmacogenet-
crease during the acute phase responses.
105. Tom`as M, Sent`ı M, Garcia-Faria F, et al.
115. Gonzalvo MC, Gil F, Hernandez AF, et al.
paraoxonase activity and related lipopro-
teins in familial hypercholesterolemic pa-
tients. Arterioscler. Thromb. Vasc. Biol.
to EDTA, metals and mercurials. Chem.
106. Leviev I, James R. 2000. Simvastatin in-
116. Cole TB, Li WF, Richter RJ, et al.
by heavy metals. Toxicologist 66(1-S):
117. Weitman SD, Vodicnick MJ, Lech TJ.
1983. Influence of pregnancy on parathion
toxicity and disposition. Toxicol. Appl.
onase activity: a diet-controlled random-
men. Atherosclerosis 1471:405–10
and platelet-activating factor acetylhydro-
127. Hayek T, Fuhrman B, Vaja J, et al. 1997.
lase in chronic renal failure. Clin. Chem.
Reduced progression of atherosclerosis in
apolipoprotein E-deficient mice following
in diabetes mellitus and its relationship
oxidation and aggregation. Arterioscler.
to serum lipids and lipoproteins. Arte-rioscler. Thromb. Vasc. Biol. 15:1812–18
128. Aviram M, Dornfeld L, Rosenblat M, et al.
120. Mackness MI, Walker CH, Carlson LA.
1987. Low A-esterase activity in serum of
patients with fish-eye disease. Clin. Chem.
121. Ferr´e N, Camps J, Cabr´e M, et al. 2001.
atherosclerotic apolipoprotein E-deficient
mice. Am. J. Clin. Nutr. 71:1062–76
and free radical production in rats with ex-
129. Kaplan M, Hayek T, Raz A, et al. 2001.
perimental cirrhosis. Metabolism 50:997–
122. Nishio E, Watanabe Y. 1997. Cigarette
lipid peroxidation, cellular cholesterol ac-
onase activity by modification of the en-
zyme’s free thiols. Biochem. Biophys. Res.
123. James RW, Leviev I, Righetti A. 2000.
paraoxonase 1 activity in rats. J. Nutr.
paraoxonase activity and concentration in
131. Wallace AJ, Sutherland WHF, Mann JI,
patients with coronary artery disease. Cir-
thermally stressed olive oil and safflower
124. Jarvik GP, Tsai N, McKinstry LA, et al.
2002. Vitamin C and E intake is associated
activity in patients with diabetes. Eur. J.
with increased paraoxonase activity. Arte-rioscler. Thromb. Vasc. Biol. 22:1329–33
132. Yamada Y, Takatori T, Nagao M, et al.
et al. 1998. Inhibition of arylesterase by
did not confer protection from acute sarin
aliphatic alcohols. Chem. Biol. Interact.
attack. Int. J. Legal Med. 115:82–84
Résumé du budget fédéral de 2012 Le 29 mars 2012 RÉSUMÉ DU BUDGET FÉDÉRAL DE 2012 Table des matières RÉSUMÉ DU BUDGET FÉDÉRAL DE 2012 Table des matières RÉSUMÉ DU BUDGET FÉDÉRAL DE 2012 INTRODUCTION « Nous prenons aujourd’hui des mesures ambitieuses afin de la réaliser pleinement de manière à donner l’espoir à nos enfants et à nos pet
Blue Cross & Blue Shield of Rhode Island Preferred Drug List Effective October 2011 – March 2012 Three Tier Commercial Premier Formulary Guide 500 Exchange Street Providence, RI 02903-2699 www.BCBSRI.com Blue Cross & Blue Shield of Rhode Island is an independent licensee of the Blue Cross and Blue Shield Association Preferred Drug List DRUG C
 cross-species comparisons, from animal experiments using purified PON1, andmore recently from studies with PON1 knockout mice. Earlier findings indicatedthat birds, which have no to very low plasma PON1 activity, were more sensitivethan rats to the toxicity of various OPs (30). In turn, rats were more sensitive to thetoxicity of OPs than rabbits, whose plasma PON1 activity is seven times higher(31). Although several other factors may contribute to the species differences inOP toxicity, these early findings suggested that low plasma PON1 activity wouldincrease sensitivity to the acute effects of OPs.
cross-species comparisons, from animal experiments using purified PON1, andmore recently from studies with PON1 knockout mice. Earlier findings indicatedthat birds, which have no to very low plasma PON1 activity, were more sensitivethan rats to the toxicity of various OPs (30). In turn, rats were more sensitive to thetoxicity of OPs than rabbits, whose plasma PON1 activity is seven times higher(31). Although several other factors may contribute to the species differences inOP toxicity, these early findings suggested that low plasma PON1 activity wouldincrease sensitivity to the acute effects of OPs.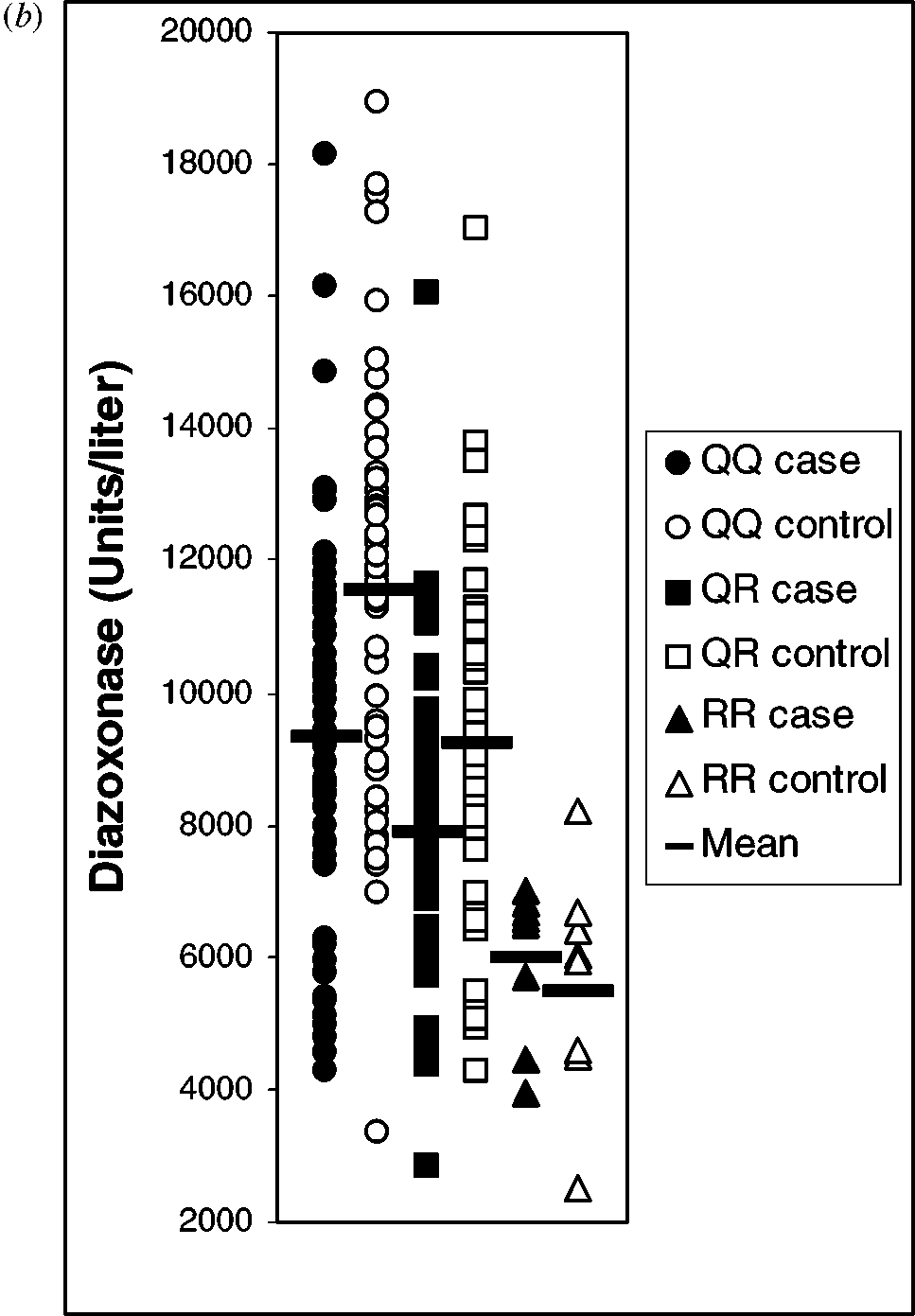 from rabbit serum (31a) was given by intravenous injection to rats (32). Adminis-tration of the enzyme raised rat serum PON1 activity toward paraoxon and chlor-pyrifos oxon by 9- and 50-fold, respectively. When rats were challenged witheither of these OPs, a significant protection [assessed by measuring inhibition ofacetylcholinesterase (AchE) in different tissues] was observed, particularly againstchlorpyrifos oxon. The protection was more prominent in two target tissues (brainand diaphragm), and was also present when OP exposure occurred by the dermalroute, as is often the case for occupationally exposed workers (32).
from rabbit serum (31a) was given by intravenous injection to rats (32). Adminis-tration of the enzyme raised rat serum PON1 activity toward paraoxon and chlor-pyrifos oxon by 9- and 50-fold, respectively. When rats were challenged witheither of these OPs, a significant protection [assessed by measuring inhibition ofacetylcholinesterase (AchE) in different tissues] was observed, particularly againstchlorpyrifos oxon. The protection was more prominent in two target tissues (brainand diaphragm), and was also present when OP exposure occurred by the dermalroute, as is often the case for occupationally exposed workers (32).