Ordered, immediately called back and the same day delivered the order.Very pleased with the work. Thank you for prompt and accurate work https://africarx.co.za/buy-levitra-south-africa.html great prices, delivered on the day of the order. Pleasant managers consult by phone.
Matthias-lange.eu
MOLECULAR INFORMATION FUSION FOR METABOLIC NETWORKS
RALF HOFESTÄDT, MATTHIAS LANGE AND UWE SCHOLZ
Bioinformatics / Medical Informatics Institute of Technical and Business Information Systems Otto-von-Guericke-University Magdeburg P.O. Box 41 20, 39016 Magdeburg, Germany E-mail: {hofestae|mlange|uscholz}@iti.cs.uni-magdeburg.de Tel.: ++49-391-67-18659 Fax: ++49-391-67-12020
Today molecular information systems are available that integrate different molecular database systems. However, the electronic information system KEGG represents the Biochemical Pathways and allows the access to different database systems which show the static representation of the molecular data and knowledge. The next important step is to implement molecular information systems which will allow to integrate different molecular database systems and analysis tools. In our paper we present an Integrative Molecular Information System for the simulation of metabolic networks.
1 Introduction
The architecture of our molecular information system allows the information fusion based on different database systems. For the simulation of metabolic networks we use the kernel of our simulation environment Metabolika which enables the interactive simulation of biochemical networks [6]. The idea of the MARGBench project [4] is to connect the simulation kernel with the WWW data sources using the database integration software. Therefore, molecular knowledge can be transferred into analytical metabolic rules - the language of Metabolika. Based on that integration software the simulation of metabolic processes is available. The configuration of Metabolika is represented by the actual metabolite concentrations. Metabolika allows the calculation of all possible configurations (derivation tree) based on the selected metabolic knowledge (biochemical scenario) and the start configuration.
Modelling of Metabolic Networks
The availability of the rapidly increasing volume of molecular data on genes, proteins and metabolic pathways enhances our capability to study cell behaviour. To understand the molecular logic of cells, we must be able to analyse metabolic processes and gene networks in qualitative and quantitative terms. Therefore, modelling and simulation are important methods.
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
The kernel of our system represents an inference mechanism which can be interpreted as a rule based system [6]. Our model is an extension of the Chomsky type-0 grammar. Defining a global rule, this formalisation allows the representation of genetic, biosynthetic, and cell communication processes. Using abstract concentration rates (integer values), we expand this discrete model. This has been realised by using multi-sets for the representation of metabolites. Metabolites are molecular structures or substance concentrations. Furthermore enzymes are proteins which catalyse biochemical reactions, whereas inducers and repressors are metabolites which are able to accelerate or slow down (prevent) biochemical reactions.
In our model the biochemical concentration of a cell is a mixture of these
components. By these definitions the abstract metabolism is given by the actual cell state (configuration) and the specific metabolic reaction rules. Therefore, the basic unit of our system is the metabolic rule. This is a formal construction which is able to describe different metabolic reactions. In that chapter we will present the basic structure of that rule based system.
Regarding a biochemical reaction, we identify the following situation. A
substance or a concentration (S) will be transferred into a product or a concentration (P). This metabolic reaction can be influenced by a concentration of inhibitor metabolites (I) or/and a concentration of enhancer metabolites (E). Using formal languages and the definition of grammars, the substance or the substance concentration can be interpreted as the left side of the rule and the metabolic product as the right side of the metabolic rule (Figure 1).
Figure 1. Metabolic Rule: S (substrate), P (product), I (inhibitor), E (enhancer) and p (rule probability)
Moreover, the inhibitors will reduce and the enhancers will increase the flux.
The actual influence of I and E will depend on the concentration of that elements regarding their actual configuration. Furthermore, each element of the set I and E represents a specific function which consists of two parameters. The first parameter is the specific threshold of the metabolite and the second parameter is his reaction
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
behaviour (kinetics). Regarding the reaction behaviour, the user of our simulation environment can choose between three different functions: hyperbolic, sigmoid, and linear. The literature shows that metabolites are characterised by their specific reaction behaviour. However, the sigmoid and hyperbolic behaviour seem to be quite common.
A 5-tuple (S, P, E, I, p) with p ∈[0,1]Q and the multi-sets S,P,E,I is called a
metabolic rule. p is called rule probability, S (substance) a set of preconditions, P (product) a set of post conditions, E (enhancer) a set of catalysed conditions, and I (inhibitor) a set of inhibitor conditions.
Based on the metabolic rule we are able to define the basic model. G=(Z,R,z0) is
called metabolic system. Z is a set of configurations, z0 is called start configuration, and R is a set of metabolic rules which is called metabolic rule set.
The first step to use the rule based model is the representation of the metabolic
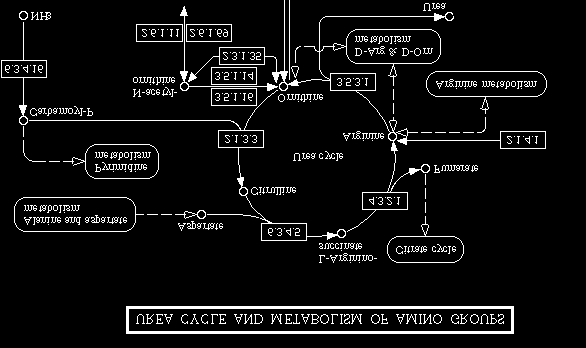
knowledge by using the specific rule based systems. In the case of biochemical reactions we can translate metabolic pathways directly. Regarding a graphical representation of metabolic pathways [11], every edge (sub graph) will represent a metabolic rule as follows: the left node (father) of this sub graph, where the edge will go out, is the S multi-set, the right node will present the P multi-set. Enhancers and inhibitors can be found by circles or numbers pointed to that edge.
Figure 2. Part of the urea cycle from the KEGG system
Using our prototype BioBench, this metabolic pathway, which is shown in
Figure 2, can be picked up from the KEGG system [12] located in Japan and will be translated directly into the language of Metabolika. The following example illustrates the translation process of the urea cycle from the KEGG-system into the
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
language of the simulation tool. In this case the EC numbers are replaced by the full enzyme names (e. g. 2.1.3.3 is replaced by Ornithine carbamoyltransferase).
• rule r1: ({Carbamoyl-Phosphate, Ornithine}, {Citrulline},
{Ornithine carbamoyltransferase}, ∅, 1.0)
• rule r2: ({Citrulline, Aspartate}, {L-Arginino-succinate},
{Argininosuccinate synthase}, ∅, 1.0)
• rule r3: ({L-Arginino-succinate}, {Fumarate, Arginine},
{Argininosuccinate lyase}, ∅, 1.0)
• rule r4: ({Arginine}, {Ornithine, Urea}, {Arginase}, ∅, 1.0)
Moreover, regarding any biochemical reactions we can discuss the processes of
gene regulation (micro-pathways) and the processes of cell communications. Therefore, the BioBench server allows the access to the TRANSFAC database [5].
Figure 3. Model of the gene regulation process
The following rule set shows how the gene regulation process (see Figure 3) can
be translated into the language of Metabolika:
• rule ra: (∅, {Repressor}, {RNA-Polymerase, ATP, P1}, ∅, p)
• rule rb: ({A}, {Inductor}, ∅, ∅, p)
• rule rc: ({Inductor, Repressor}, {Inductor-Repressor}, ∅, ∅, p)
• rule rd: ({Inductor-Repressor}, {E1,E2}, {RNA-Polymerase, ATP, P2}, ∅, p)
• rule re: ({A}, {A'}, {E1}, ∅, p)
• rule rf: ({A'}, {A''}, {E2}, ∅, p) HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
Using metabolic rules, the modelling of cell communication processes (see
Figure 4) is simple. Metabolites will go into (will leave) the cell, if only the P (S) component of that rule is not empty. However, the inhibitor and enhancer component of the metabolic rule allows the simulation of receptor effects.
Figure 4. The abstract cell communication process
• rule ra: ({s}, ∅, ∅, ∅, 1.0)
• rule rb: (∅, {p}, ∅, ∅, 1.0)
Therefore, metabolic networks will be represented by a set of metabolic rules.
The processing mechanism of our model is as follows. Any rule is called activated, if the elements (concentrations) of the S component are elements (concentrations) of the actual configuration. Any activated rule r can go into action. The action of r will modify the actual cellular state of the metabolic system. All elements of the actual cellular state, which are elements of the substance set (S) of rule r, will be eliminated. All elements of the product component (P) will be added. Therefore, the action of rule r is a substitution P for S which can produce a new configuration.
Example: Consider the rule set of the Urea pathway and let ( Carbamoyl-
Phosphate, Ornithine, Ornithine carbamoyltransferase) be the actual cell state. However, only the first rule is activated and will go into action, because the probability value is 1.0. The action of that rule will consume the Carbamoyl-Phosphate and Ornithine molecules and will produce a Citrulline molecule. This biochemical reaction will be catalysed by the enzyme Ornithinecarbamoyltransferase.
The one step derivation of a metabolic system is defined by the (quasi)
simultaneous action of all activated rules. Therefore, we consider the set of all activated rules and determine two new sets: the before-set and the after-set. The before-set includes all before elements of the activated rules. A definition of the after-set is analogous. Using these sets, the one-step derivation could be interpreted as an addition and subtraction of concentrations.
Example: Regarding again the rule set of the urea pathway, and let the actual
cell state be ( Carbamoyl-Phosphate, Ornithine, Ornithine carbamoyltransferase,
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54 Citrulline, Aspartate, Argininosuccinate synthase, Arginine, Arginase). However, the first (r1) , second (r2) and last rule (r4) are activated. Therefore, different one-step-derivations can be produced (non-deterministic rule system).
Each action can be interpreted as an independent event. Therefore, the
probability of each one-step derivation can be calculated from the absolute probability values of all activated and deactivated rules. In our simulation system this will be done by multiplying these values. One-step derivations inductively produce complex derivation trees of configurations.
Based on the theory of the rule based modelling of metabolic processes we
developed the simulation shell Metabolika [6]. Metabolika allows the integrative simulation of biochemical networks including cell communication processes. Metabolika is implemented in C and runs on a SUN Sparc workstation. Its main parts are the rule editor and the configuration editor/browser.
The set of the reachable configurations is an infinite set, and the set of all derivations is enumerable. Moreover, the set of all configurations is not decidable [7]. Use of concentrations (multi-sets) is the main reason for the indecidability. However, this result implies that no interesting question can be solved in the research field of biotechnology.
In practice biochemical systems are restricted. In our model we can restrict the
depth and width of the derivation process. Therefore, important questions are decidable, and we have to discuss the complexity of the derivation algorithm. If we restrict the derivation depth the language L(G, i) is decidable. L(G, i) is the set of all configurations which can be produced from the start configuration by the application of up to i derivation steps. Hence for a metabolic system with the generation depth i it is decidable, if k is a member of L(G, i). Based on the exponential complexity of the derivation process, this question cannot be solved in practice if i is high. Therefore the calculation of a derivation tree is not possible in practice. However, using our simulation tool we have to restrict the derivation depth and/or width.
Information Fusion
The presence of numerous informational and programming resources on gene networks, metabolic processes, gene expression regulation, etc., described above, raises an acute problem of data integration and suitable access. Goal of such integration is to create a virtual informational environment, enabling an access to the significant information on the basis of simultaneous exploration of many databases available via Internet. Effective possibilities for data base integration are provided by the World Wide Web technology.
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
One of the most developed technologies of WWW integration of molecular
databases uses the Sequence Retrieval System (SRS). It is based on local copies of each component database, which have to be provided in a text-based format. The results of the query are sets of WWW-links. Thus the user can navigate through these links. Up to now, several hundreds of databases on molecular biology are integrated under SRS [3]. However, within the frames of this approach, data fusion is still a task of the user. We also do not find real data fusion; i. e. data for one real world object (e. g. an enzyme) coming from two different databases (e. g. KEGG and BRENDA [14]) is represented two times by different WWW page objects.
Therefore, research groups try to integrate molecular databases on a higher
level than the SRS approach. For, they apply results of current database research, e. g. federated database systems, data warehousing architectures or data mining techniques [1]. Many bioinformatics problems require
1. access to data sources that are high in volume, highly heterogeneous and
complex, constantly evolving and geographically dispersed,
2. solutions that involve multiple carefully sequenced steps, 3. information to be passed smoothly between the steps, 4. increasing amount of computation and 5. increasing amount of visualisation.
BioKleisli (see [2]) is an advanced technology designed to handle the first three
requirements directly. In particular, BioKleisli provides the high-level query language CPL that can be used to express complicated transformation across multiple data sources in a clear and simple way. In addition, while BioKleisli does not handle the last two requirements directly, it is capable of distributing computation to appropriate servers and initiating visualisation programs. The idea of our project is to present a virtual laboratory for the analysis of molecular processes (diseases). Therefore, we integrated different specific database systems which represent molecular and medical knowledge and a simulation environment (see [9, 4] for more information).
The Biomedical WorkBench
As mentioned before, the analysis of existing systems shows that on the one hand many database systems which contain data about biochemical reactions are available. On the other hand powerful analysis tools e.g. simulation tools in this domain exist.
We built an integrated molecular information system, which is called
Biomedical Workbench (BioBench) [9]. The idea of this system is to present a virtual laboratory for the analysis of molecular processes. For that reason we integrated different database systems which represent molecular and medical
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
knowledge. We called this integration information fusion. Hence we have the possibility to detect equal data in various databases. The graphical user interface gives the user access to a compact local information system. In case of modelling and simulation of metabolic processes the specific biochemical knowledge will be identified by using these database systems. As next step, this knowledge will be transferred automatically into the language of analytical metabolic rules, the language of Metabolika. The simulation of this biochemical reactions will be produced by the kernel of Metabolika and results of this simulation will be visualised by a special visualisation component (VisTool).
The BioBench prototype integrates three different databases: KEGG (see [12]),
MDDB (see [8]) and parts of the TRANSFAC database (see [5]). This integration is based on a fix and hard implementation of special adapters for the access onto the different systems. These adapters transferred the data of the component systems in a unique form. The basis for the unique representation form is a global data model which integrates the models of the component systems. Figure 5 shows the architecture of the prototype.
Graphical User Interface(runs on a WWW-Client)
(M etabolic Di s e a s e s D atab a s e, Figure 5. Architecture of the BioBench prototype
So far, the main application of the BioBench prototype was the analysis of
metabolic pathways. In one component database system, the MDDB, information about metabolic diseases are stored. These diseases are caused by genetic defects. The result is, that a special enzyme for metabolism can not be synthesised. At the
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
end a special biochemical reaction could not be catalysed by the missing enzyme and substrates could not be transferred into products. With help of BioBench and the including simulation tool it is possible to analyse this break in a metabolic pathway and to look for alternative pathways. Furthermore, based on the BioBench prototype, we are developing a new system which is called MARGBench.
Summarising, the biggest advantage of this BioBench is its possibility to
automatically generate the input data for the simulation tool. It is not necessary for the user to type in the simulation parameters manually. Nevertheless, the fixed architecture of the systems is a disadvantage. The adapter are fix implemented software modules. If the access interface of any component database will be modified, then a corresponding adapter must be newly implemented. Moreover, only one simulation tool is fix interlinked.
Logical Integration of Molecular Databases
In the field of molecular biology a steady stream of data is generated and researchers are collecting their knowledge in fast growing databases. These databases are distributed around the world. Several of them are accessible through the Internet which is the basis for a wide and public use. For the adequate work of biologists a wide range of this molecular-biological data are essential. For this reason it is necessary to look at as many databases as possible. That raises the question, how to obtain that data.
First, the manual way is to interactively browse the databases. Hereby it is not
really possible to make the data available to any computer program. This approach is useful for investigation, because the effort of getting these data as an input into a computer program, e.g. via "cut and paste" or typewriting is not very workable.
Another approach is to realise a computer supported data access. Therefore, an
additional software layer is required, that provides the mechanism of access and delivery of merged data. Thus the biological tools access the several databases not directly, rather they get the data from a special software layer which is called the Database Integration Server. Hence the following problems are to solve:
• solving the several kinds of database heterogeneity
• transparent physical database access
To fulfil the above requirements four approaches are proposed [10]:
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
1. Hypertext navigation (see [3]) 2. Data warehouse (see [13]) 3. Multi database queries (see [2]) 4. Federated databases (see [1] )
In correlation with the research project Modelling and Animation of Regulative Gene Networks an architecture has been designed which realises a logical database integration based on the concept of federated databases. The system architecture (see WWW address: http://wwwiti.cs.uni-magdeburg.de/~mlange/BioDataServer/) has been implemented as a prototype called BioDataServer [4].
A workable Internet access to the molecular-biological databases is the main
requisition for a database integration. Thereby several problems are to solve:
• different interfaces (e.g. CGI, JDBC)
• different query languages (SQL, OQL, non-standardised)
• different data presentations (HTML, flat files, database objects)
• different data structures (static, dynamic)
To hide the access heterogeneity, the BioDataServer uses adapter for the
physical data access. For each data source a special adapter exists which is able to handle the data retrieval. In the case of an HTML-based data source the adapter accesses the specific URL and parses the resulting HTML-page. Current work studies the possibilities of semi-automatic generation of adapters (see [4]).
To obtain a complete and wide spectrum of data it is recommendable to access
as many databases as possible. Therefore the queries will be distributed to each relevant database. The information how to distribute the queries are stored in an integrated user scheme. This scheme is relational and defines the source for each attribute. On the basis of these schemes the BioDataServer accesses the related attributes at the specific databases.
Furthermore an automatic mechanism for merging the attributes from the
various databases is necessary. This is the task of the integration layer of the BioDataServer and can be solved, using mathematical set operations.
The premise to access the Database Integration Server by computer programs is
the definition of an interface. Because the server should be accessible through the Internet, a communication protocol and a query language must be specified. Nowadays a lot of database systems exist which support SQL as the query language, which in turn is based on the relation model and standardised. Nearly all commercial database systems support SQL and thereby it has been established worldwide. This was the reason to support SQL by the BioDataServer. In the field of interfaces for remote database access, different techniques have been established e.g. JDBC and ODBC. ODBC is currently only supported by Microsoft platforms. Therefore the BioDataServer offers a JDBC driver, which provides a standardised database access
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
to JAVA applications. Consequently any JAVA platform can simply access the BioDataServer by related JAVA programs.
To fulfil extended requirements to a universally applicable database integration
server, a new architecture was developed and has been implemented as a JAVA application. The main advantages of this BioDataServer are
• the transparent physical database access,
• dynamic building of a new virtual, logical integrated database
Summarising, this kind of database integration is a step for the standardised
integration of worldwide distributed molecular databases and the related software tools.
Summary and Outlook
First, a short description of the current situation in the field of bioinformatics was given in this paper. On the one hand, a huge amount of data in heterogeneous systems is available. On the other hand, more and more powerful analysis tools are growing up. One main research interest is the integration of the databases and the analysis tools.
Following this line, we illustrated the theory of our rule based approach which
is implemented in the Metabolika system. An example showed the simulation possibilities of this tool. Furthermore, the idea of information fusion and our prototype BioBench were described. Finally an approach for the logical integration of molecular databases was presented in a detailed form and some advantages of this integration were illustrated.
The most frequently introduced concepts of information fusion are implemented
in our MARGBench prototype. During our current work we are connecting the pieces of our system. After voluminous and intensive tests we plan to bring our system in the WWW. Some more information about the prototype and our project is available under the address: http://wwwiti.cs.uni-magdeburg.de/iti_bm/marg/.
7 Acknowledgements
This work is supported by the German Research Council (DFG).
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54 References
1. Conrad S., Federated Database Systems: Concepts of Data Integration.
Springer-Verlag, Berlin/Heidelberg, 1997. (In German).
2. Davidson S. B., Overton C., Tannen V. and Wong L., BioKleisli: a digital
library for biomedical researchers. International Journal on Digital Libraries, 1:36-53, 1997.
3. Etzold T., Ulyanow A. and Argos P., SRS: Information Retrieval System for
Molecular Biology Data Banks. Methods in Enzymology, 266:114-128, 1996.
4. Freier A., Hofestädt R., Lange M. and Scholz U., Integration, Modellierung und Simulation metabolischer Wirknetze. Preprint 13, Fakultät für Informatik, Universität Magdeburg, 1999. (In German).
5. Heinemeyer T., Chen X., Karas H., Kel A. E. , Kel O. V., Liebich I., Meinhardt
T., Reuter I., Schacherer F. and Wingender E., Expanding the TRANSFAC database towards an expert system of regulatory molecular mechanisms. Nucleid Acids Research, 27(1):318-322, 1999.
6. Hofestädt R. and Meinecke F., Interactive Modelling and Simulation of
Biochemical Networks. Computers in Biology and Medicine, 25(3):321-334, 1995.
7. Hofestädt R., Theorie der regelbasierten Modellierung des Zellstoffwechsels.
Aachen: Shaker, 1996. (In German).
8. Hofestädt R., Prüß M., Scholz U. and Urban H., The Metabolic Diseases
Database (MDDB) - A Molecular Database Toolkit for the Detection of Inborn Errors. In O. Zimmermann and D. Schomburg, editors, Proceedings of the German Conference on Bioinformatics (GCB '98), Köln, October 7-10, 1998.
9. Hofestädt R. and Scholz U., Information Processing for the Analysis of
Metabolic Pathways and Inborn Errors. BioSystems, 47(1-2):91-102, 1998.
10. Karp P. D., A Strategy for Database Interoperation. Journal of Computational
11. Michal G., Biochemical Pathways. Heidelberg: Spektrum Akademischer
12. Ogata H., Goto S., Kazushige S., Fujibuchi W., Bono H. and Kanehisa M.,
KEGG: Kyoto Enzyclopedia of Genes and Genomes. Nucleid Acids Research, 27(1):29-34, 1999.
13. Ritter O., Comprehensive Genome Information Systems. In S. Suhai, editor,
Theoretical and Computational Methods in Genome Research, pages 177-184. New York et al: Plenum Press, 1997.
14. Schomburg D., Schomburg I., Chang A. and Bänsch C., BRENDA the
Information System for Enzymes and Metabolic Information. In R. Giegerich, R. Hofestädt, T. Lengauer, W. Mewes, D. Schomburg, M. Vingron, and E. Wingender, editors, Proceedings of the German Conference on Bioinformatics (GCB '99), Hannover, October 4-6, pages 226-227, 1999.
HofLanSch.doc submitted toWorld Scientific 04.07.00 : 14:54
I diuretici trovano applicazione clinica in varie situazioni tra cui spiccano:• la risoluzione degli edemi derivanti Cirrosi epatica Ostruzione Ridotta sintesi Aumento pressione plasmaproteine sanguigna Meccanismo di formazione degli edemi nella cirrosi epatica Danno renale Proteinuria Diminuzione Ridotta volemia: press. oncotica aumento aldosterone Mecc
Elenco Pubblicazioni in extenso di Enrico Aitini e Colleghi Struttura Complessa di Oncologia Medica ed Ematologia (autore, co-autore, principal investigator, main co-investigator) Franciosi V, Barbieri R, Aitini E et al: GEMCITABINE AND OXALIPLATIN: A SAFE AND ACTIVE REGIMEN IN POOR PROGNOSIS ADVANCED NON-SMALL CELL LUNG CANCER PATIENTS. Lung Cancer. 2003 Jul;41(1):101-6 Gridelli C, Gallo
 behaviour (kinetics). Regarding the reaction behaviour, the user of our simulation environment can choose between three different functions: hyperbolic, sigmoid, and linear. The literature shows that metabolites are characterised by their specific reaction behaviour. However, the sigmoid and hyperbolic behaviour seem to be quite common.
A 5-tuple (S, P, E, I, p) with p ∈ [0,1]Q and the multi-sets S,P,E,I is called a
metabolic rule. p is called rule probability, S (substance) a set of preconditions, P (product) a set of post conditions, E (enhancer) a set of catalysed conditions, and I (inhibitor) a set of inhibitor conditions.
Based on the metabolic rule we are able to define the basic model. G=(Z,R,z0) is
called metabolic system. Z is a set of configurations, z0 is called start configuration, and R is a set of metabolic rules which is called metabolic rule set.
The first step to use the rule based model is the representation of the metabolic
knowledge by using the specific rule based systems. In the case of biochemical reactions we can translate metabolic pathways directly. Regarding a graphical representation of metabolic pathways [11], every edge (sub graph) will represent a metabolic rule as follows: the left node (father) of this sub graph, where the edge will go out, is the S multi-set, the right node will present the P multi-set. Enhancers and inhibitors can be found by circles or numbers pointed to that edge.
Figure 2. Part of the urea cycle from the KEGG system
behaviour (kinetics). Regarding the reaction behaviour, the user of our simulation environment can choose between three different functions: hyperbolic, sigmoid, and linear. The literature shows that metabolites are characterised by their specific reaction behaviour. However, the sigmoid and hyperbolic behaviour seem to be quite common.
A 5-tuple (S, P, E, I, p) with p ∈ [0,1]Q and the multi-sets S,P,E,I is called a
metabolic rule. p is called rule probability, S (substance) a set of preconditions, P (product) a set of post conditions, E (enhancer) a set of catalysed conditions, and I (inhibitor) a set of inhibitor conditions.
Based on the metabolic rule we are able to define the basic model. G=(Z,R,z0) is
called metabolic system. Z is a set of configurations, z0 is called start configuration, and R is a set of metabolic rules which is called metabolic rule set.
The first step to use the rule based model is the representation of the metabolic
knowledge by using the specific rule based systems. In the case of biochemical reactions we can translate metabolic pathways directly. Regarding a graphical representation of metabolic pathways [11], every edge (sub graph) will represent a metabolic rule as follows: the left node (father) of this sub graph, where the edge will go out, is the S multi-set, the right node will present the P multi-set. Enhancers and inhibitors can be found by circles or numbers pointed to that edge.
Figure 2. Part of the urea cycle from the KEGG system